






Jueves
3 de Julio de 2025

22 de abril de 2020
Un estudio genético reconstruye el camino evolutivo del SARS-CoV-2 desde China a Europa y América del Norte, revelando tres variantes distintas del virus desde los orígenes de la pandemia.
Utilizando las mismas técnicas que se emplean para mapear los movimientos de las poblaciones humanas prehistóricas a través del análisis de mutaciones de ADN, un equipo de científicos de diversas instituciones del Reino Unido y Alemania ha reconstruido los primeros movimientos de la infección por SARS-CoV-2 desde su epicentro en la ciudad china de Wuhan hacia el resto del mundo. El resultado, una especie de instantánea sobre los orígenes de la pandemia, se ha publicado en la revista Proceedings of the National Academy of Sciences (PNAS).
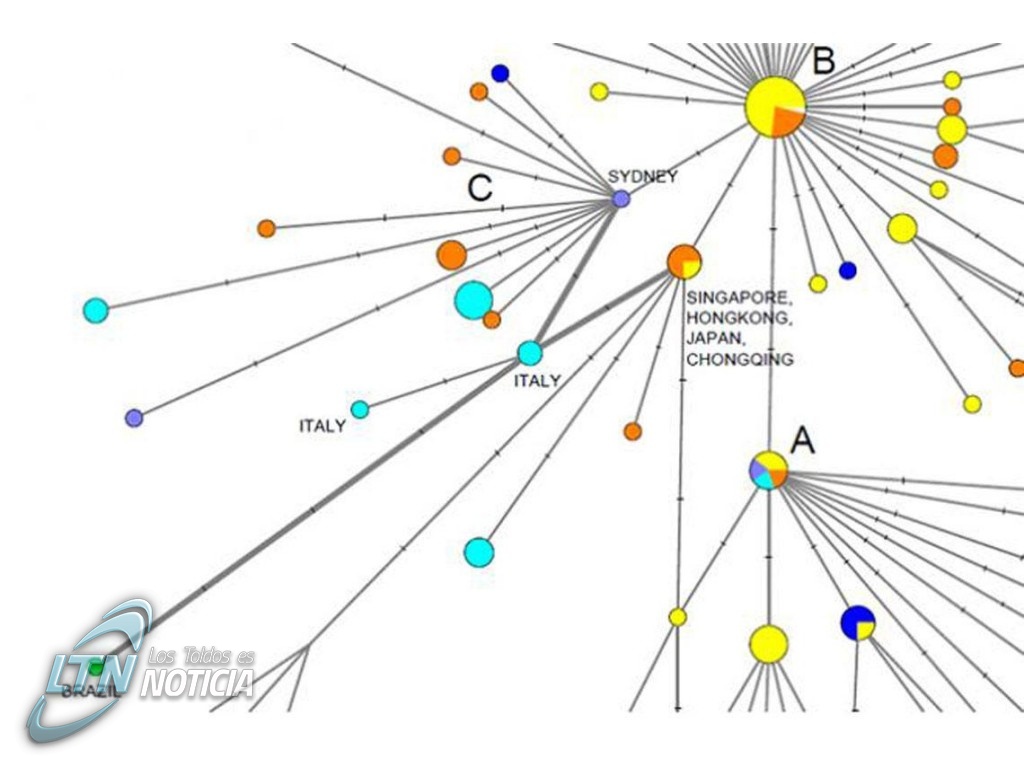
Los científicos utilizaron los primeros 160 genomas completos del virus que se secuenciaron a partir de pacientes humanos para reproducir parte de la propagación inicial del nuevo coronavirus a través de sus mutaciones. En concreto, los datos de los genomas fueron recogidos de personas de todo el mundo entre el 24 de diciembre de 2019 y el 4 de marzo de 2020. Los resultados revelaron tres variantes distintas del virus, que en realidad serían grupos formados por linajes estrechamente relacionados y que denominaron ‘A’, ‘B’ y ‘C’.
Una de las primeras sorpresas encontradas fue que el tipo de coronavirus más parecido al que se descubrió en los murciélagos y pangolines (el tipo ‘A’), estaba presente en Wuhan, pero no era el más abundante. Esta variante ha sido identificada en pacientes chinos y en estadounidenses residentes en Wuhan. Además, se encontraron versiones mutadas de ‘A’ en otros pacientes de EE UU y Australia.
La variante ‘B’ habría sido la más prevalente en el este de Asia, pero no llegó mucho más allá, algo que según los investigadores se podría deberse a dos motivos: uno sería un fenómeno que en evolución se conoce como ‘efecto fundador’. Se trata de un cuello de botella genético que ocurre cuando se establece un nuevo tipo de virus a partir de un pequeño grupo aislado de infecciones.
Otra explicación que, según los autores, valdría la pena considerar, se refiere al desarrollo de algún tipo de resistencia contra esta variante de la infección fuera de Asia oriental. “Parece que, en esta fase inicial, vemos una tasa de mutación más lenta en el este de Asia que en otros lugares”, explica Peter Forster, investigador en la Universidad de Cambridge y autor principal del trabajo. “El virus de tipo ‘B’ podría adaptarse inmunológica o ambientalmente a una gran parte de la población de Asia oriental, pero es posible que deba mutar para superar esa resistencia fuera de dicha región”, argumenta.
La variante europea
El coronavirus denominado ‘tipo C’ habría sido el más abundante en Europa, al menos en los inicios de la infección, y esta variante fue aislada en los primeros casos detectados en Francia, Italia, Suecia e Inglaterra. También ha sido encontrada en pacientes de Singapur, China, Hong Kong y Corea del Sur, pero no así en la muestra de China continental.
Los investigadores destacan que estas técnicas de análisis filogenético pueden ser útiles para rastrear posibles rutas de infección y aplicarse a las últimas secuenciaciones del genoma del coronavirus para ayudar a predecir los futuros puntos calientes donde la infección podría volver a estallar para contenerla adecuadamente.
También hay que tener en cuenta que este análisis se hizo con los datos obtenidos justo al comienzo del estallido de la COVID-19. “La red viral que hemos detallado en el artículo es tan solo una instantánea de las primeras etapas de la epidemia, antes de que todos los cambios evolutivos sufridos por el virus se oscurecieran debido al gran número de mutaciones”, explica Forster. “Es como atrapar una supernova justo al comienzo de la explosión”, concluye.
3 de Julio de 2025

Continuos problemas te asaltarán durante la jornada de hoy al trasladarte a tu lugar de trabajo. Paciencia y calma. Lograrás vislumbrar que los sentimientos de tu pareja no son tan genuinos como pensabas. Aclara esto de inmediato. Cultiva tus capacidades en cada oportunidad que tengas. Esto te permitirá aprovechar las oportunidades a medida que aparezcan.Sugerencia: Delinea los limites de ciertas actitudes nocivas para la pareja de manera clara y concisa. No caigas en ambigüedades o los problemas te asaltarán.
| OFICIAL COMPRA | OFICIAL VENTA |
|---|
 Email: [email protected]
Email: [email protected] En Twitter: @ltnoticia
En Twitter: @ltnoticia
 En Facebook: Los toldos es noticia Comunidad ( Y vos mismo te das el alta)
En Facebook: Los toldos es noticia Comunidad ( Y vos mismo te das el alta)